Publishing eDNA Data in OBIS
From sequences to a global open biodiversity record
2026-07-09
eDNA data in OBIS

- OBIS currently holds 128 datasets with eDNA-derived occurrences, including 44,5 million records
- About 2 million distinct sequences
- That is 2% of all datasets, but 25% of all occurrence records in OBIS
The eDNA Metabarcoding Workflow

Figure adapted from NatureMetrics and Gill et al. 2016 · Emilie Boulanger
Darwin Core + DNA Derived Data Extension
- Darwin Core: shared vocabulary for biodiversity data
- DNA Extension: adds MIxS terms for sequences, primers, methods
- Developed jointly by GBIF, OBIS, and the genomics community
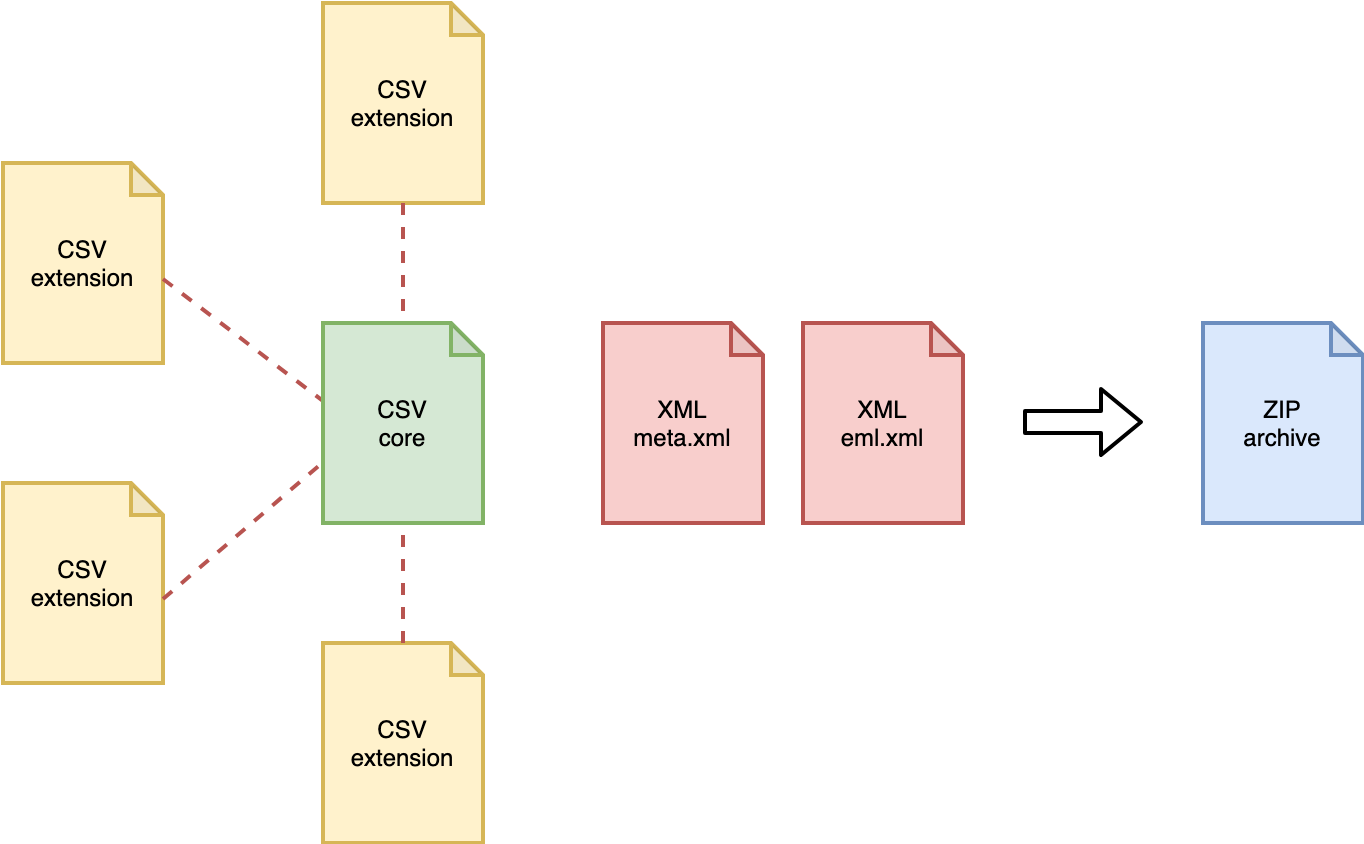
meta.xml ← links all tables
eml.xml ← metadata (who, what, where, when, how)
occurrence.csv ← the occurrence records
dna_derived.csv ← the DNA extension
The DNA publishing guide
Publishing DNA-derived data through biodiversity data platforms


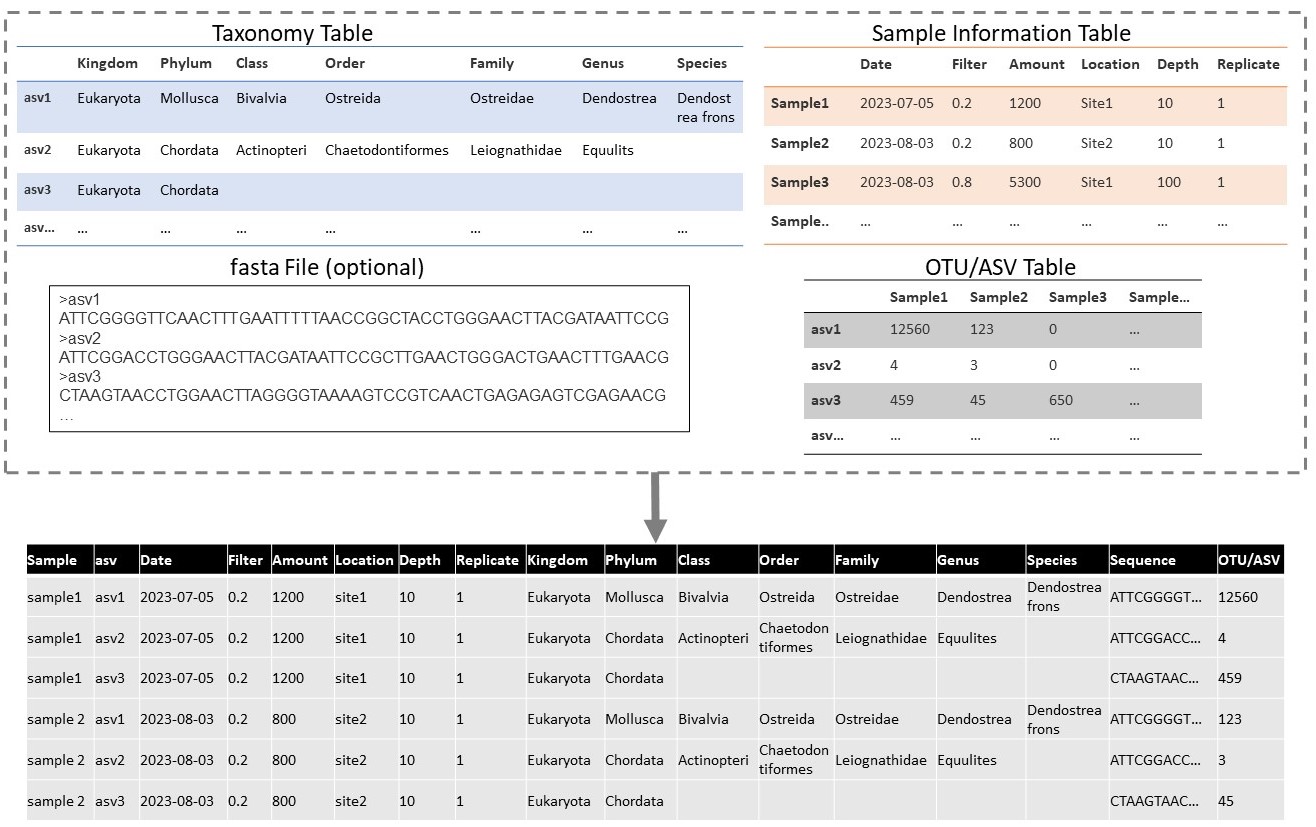
Example transformation
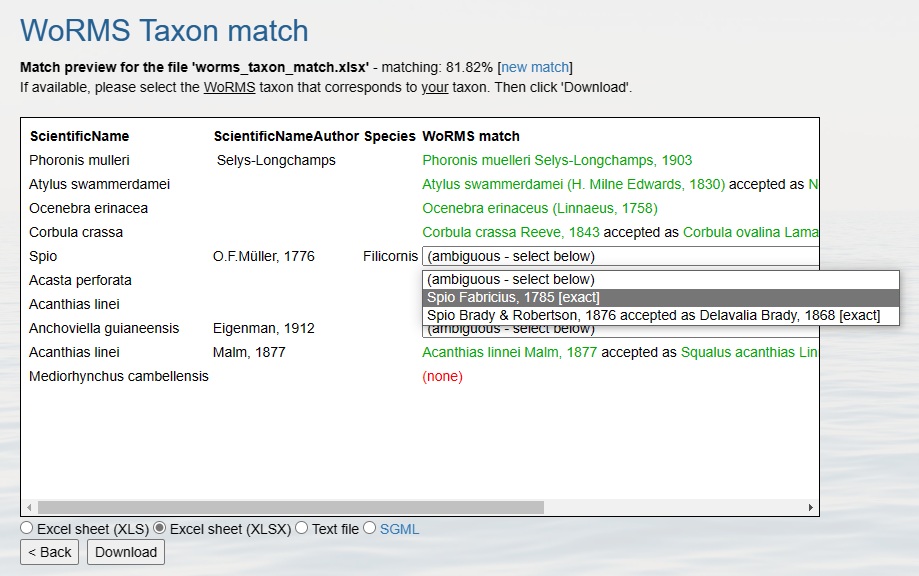
WoRMS Taxon Matching
Options for taxon matching:
- WoRMS Taxon Match Tool
- Programmatically:
No WoRMS match?
Use highest-rank taxon with a match (e.g. family or order). Keep the sequence — it can be re-identified later as reference databases improve.
Key fields to include: DNA Extension
DNA Extension (example key fields)
DNA_sequence— ASV/OTU sequencetarget_gene— e.g. “COI”, “18S rRNA”pcr_primer_name_forward— and reverseseq_meth— e.g. “Illumina MiSeq”otu_class_appr— e.g. “DADA2 v1.18”otu_db— reference databasesop— link to your protocol
 https://manual.obis.org/dna_data.html#quick-start-guide
https://manual.obis.org/dna_data.html#quick-start-guide
Example usage of Event core structure
- Why Event Core? Sample-level metadata (location, date, method) is recorded once per sampling event rather than repeated in every row.
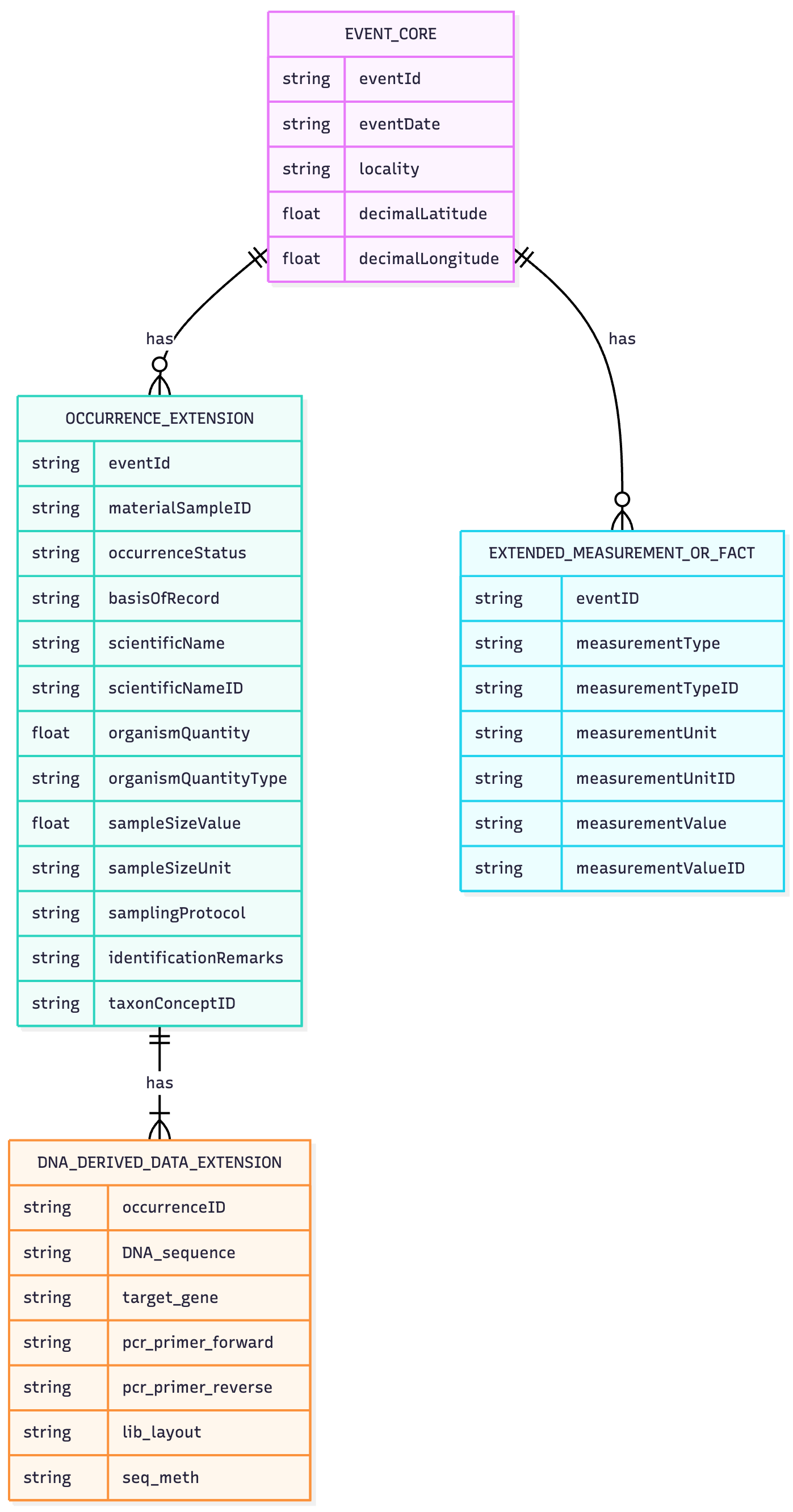
Controlled Vocabulary
Identifiers for each eMoF column
- measurementUnitID
- measurementValueID
- non-numeric values
- measurementTypeID
→ Facilitates data understanding
→ Enables data aggregation
→ Decreases misuse potential
What should vocabulary terms capture? 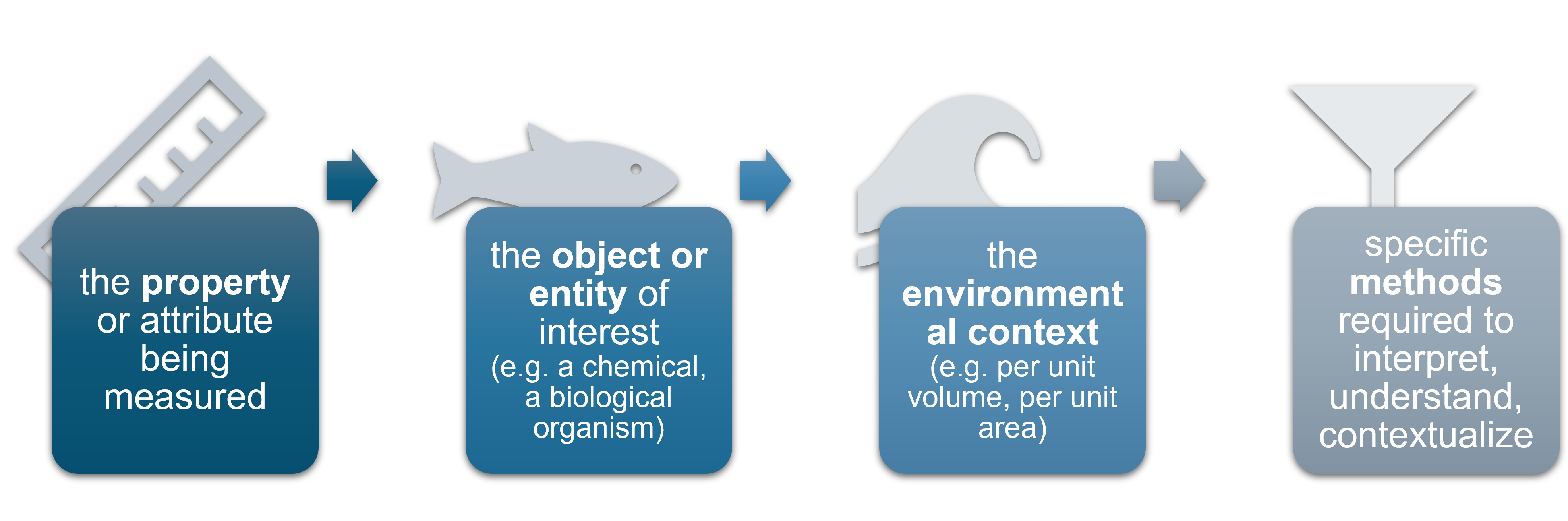
OBIS recommends using NERC vocabulary terms
